class: center, middle, inverse, title-slide .title[ # Data I/O ] .subtitle[ ## Reading and writing biological data ] .author[ ### Matt Black, Guillaume Falmagne, Scott Wolf, Diogo Melo ] .date[ ### <br> Sept. 23th, 2024 ] --- # Basic data types - Tabular - Most common type in ecology. - Usually goes into a data.frame (in R). - Excel files, csv, tsv, ... - But also yaml, json, pickle (in python), npy/npz (numpy), ... - Genomic data - Most common in genetics, bioinformatics - Usually pretty large, so requires some care and specialized data structures - Genotype data, sequence data, aligned sequence data, expression data - Databases - Relational data that is more complex than a single table --- # Tabular data ```r head(iris) ``` ``` ## Sepal.Length Sepal.Width Petal.Length Petal.Width Species ## 1 5.1 3.5 1.4 0.2 setosa ## 2 4.9 3.0 1.4 0.2 setosa ## 3 4.7 3.2 1.3 0.2 setosa ## 4 4.6 3.1 1.5 0.2 setosa ## 5 5.0 3.6 1.4 0.2 setosa ## 6 5.4 3.9 1.7 0.4 setosa ``` ```python import pandas as pd iris = pd.read_csv("data/iris.csv") print(iris.head()) ``` ``` ## Id SepalLengthCm SepalWidthCm PetalLengthCm PetalWidthCm Species ## 0 1 5.1 3.5 1.4 0.2 Iris-setosa ## 1 2 4.9 3.0 1.4 0.2 Iris-setosa ## 2 3 4.7 3.2 1.3 0.2 Iris-setosa ## 3 4 4.6 3.1 1.5 0.2 Iris-setosa ## 4 5 5.0 3.6 1.4 0.2 Iris-setosa ``` --- class: inverse, center, middle # R reading and writing --- # R: various packages to read tabular data .pull-left[ - Base functions: - Output `data.frames` - Names have `.` in them, e.g. `read.table`, `read.csv` - `readr` package - Replacements for the base functions, but with different choices - ties into the `tidyverse` packages we will see later on (super popular) - Names have `_` in them, e.g. `read_delim`, `read_csv` ] .pull-right[ - `data.table` package - Very fast because uses `fread` function; sometimes the only choice for large data - `rio` package - sort of jack of all trades magic package that attempts to figure out the correct way to read your data from the file extension - Only has one generic `import` function that mostly get's it right. ] *Lesson*: Always **check with some examples/testing** that the reading function does what you want on your file! --- # read.table .pull-left[ - General purpose table reading function. - Reads a file in table format and creates a `data.frame`. ```r # Basic Usage my_data <- read.table("data/my_file.tsv", header = TRUE, sep = "\t") head(my_data) ``` ``` ## Sepal.Length Sepal.Width Petal.Length Petal.Width Species ## 1 5.1 3.5 1.4 0.2 setosa ## 2 4.9 3.0 1.4 0.2 setosa ## 3 4.7 3.2 1.3 0.2 setosa ## 4 4.6 3.1 1.5 0.2 setosa ## 5 5.0 3.6 1.4 0.2 setosa ## 6 5.4 3.9 1.7 0.4 setosa ``` ] --- # read.csv & read.csv2 Special cases of `read.table` designed for CSV files. - `read.csv`: assumes field separator is "," and . for decimal point. - `read.csv2`: assumes field separator is ";" and , for decimal point (common in European datasets). ```r data_csv <- read.csv("my_data.csv") # For European-style CSV data_csv2 <- read.csv2("my_data_european.csv") ```  --- # readr package - Outputs a `tibble`, a kind of modern `data.frame` with some quirks - does some smart figuring-out of column types ```r # Basic Usage library(readr) my_data_tbl <- read_delim("data/my_file.tsv", delim = "\t") ``` ```r my_data_tbl ``` ``` ## # A tibble: 150 × 5 ## Sepal.Length Sepal.Width Petal.Length Petal.Width Species ## <dbl> <dbl> <dbl> <dbl> <chr> ## 1 5.1 3.5 1.4 0.2 setosa ## 2 4.9 3 1.4 0.2 setosa ## 3 4.7 3.2 1.3 0.2 setosa ## 4 4.6 3.1 1.5 0.2 setosa ## 5 5 3.6 1.4 0.2 setosa ## 6 5.4 3.9 1.7 0.4 setosa ## 7 4.6 3.4 1.4 0.3 setosa ## 8 5 3.4 1.5 0.2 setosa ## 9 4.4 2.9 1.4 0.2 setosa ## 10 4.9 3.1 1.5 0.1 setosa ## # ℹ 140 more rows ``` --- # data.table's fread - data.table is a very fast package to deal with tabular data - Has its own very particular syntax - Can deal with datasets above 100GB easily ```r library(data.table) data_dt <- fread("data/my_file.csv") data_dt ``` ``` ## Sepal.Length Sepal.Width Petal.Length Petal.Width Species ## <num> <num> <num> <num> <char> ## 1: 5.1 3.5 1.4 0.2 setosa ## 2: 4.9 3.0 1.4 0.2 setosa ## 3: 4.7 3.2 1.3 0.2 setosa ## 4: 4.6 3.1 1.5 0.2 setosa ## 5: 5.0 3.6 1.4 0.2 setosa ## --- ## 146: 6.7 3.0 5.2 2.3 virginica ## 147: 6.3 2.5 5.0 1.9 virginica ## 148: 6.5 3.0 5.2 2.0 virginica ## 149: 6.2 3.4 5.4 2.3 virginica ## 150: 5.9 3.0 5.1 1.8 virginica ``` --- # Reading data of the interwebs ```r input <- "https://raw.githubusercontent.com/Rdatatable/data.table/master/vignettes/flights14.csv" flights <- fread(input) flights ``` ``` ## year month day dep_delay arr_delay carrier origin dest air_time ## <int> <int> <int> <int> <int> <char> <char> <char> <int> ## 1: 2014 1 1 14 13 AA JFK LAX 359 ## 2: 2014 1 1 -3 13 AA JFK LAX 363 ## 3: 2014 1 1 2 9 AA JFK LAX 351 ## 4: 2014 1 1 -8 -26 AA LGA PBI 157 ## 5: 2014 1 1 2 1 AA JFK LAX 350 ## --- ## 253312: 2014 10 31 1 -30 UA LGA IAH 201 ## 253313: 2014 10 31 -5 -14 UA EWR IAH 189 ## 253314: 2014 10 31 -8 16 MQ LGA RDU 83 ## 253315: 2014 10 31 -4 15 MQ LGA DTW 75 ## 253316: 2014 10 31 -5 1 MQ LGA SDF 110 ## distance hour ## <int> <int> ## 1: 2475 9 ## 2: 2475 11 ## 3: 2475 19 ## 4: 1035 7 ## 5: 2475 13 ## --- ## 253312: 1416 14 ## 253313: 1400 8 ## 253314: 431 11 ## 253315: 502 11 ## 253316: 659 8 ``` --- # Reading generic data files ```r # Using the scan() function my_generic_file <- scan("data/my_file.csv", character()) # Using the readLines() function my_generic_file <- readLines("data/my_file.csv") my_generic_file ``` ``` ## [1] "Sepal.Length, Sepal.Width, Petal.Length, Petal.Width, Species" ## [2] "5.1, 3.5, 1.4, 0.2, setosa" ## [3] "4.9, 3, 1.4, 0.2, setosa" ## [4] "4.7, 3.2, 1.3, 0.2, setosa" ## [5] "4.6, 3.1, 1.5, 0.2, setosa" ## [6] "5, 3.6, 1.4, 0.2, setosa" ## [7] "5.4, 3.9, 1.7, 0.4, setosa" ## [8] "4.6, 3.4, 1.4, 0.3, setosa" ## [9] "5, 3.4, 1.5, 0.2, setosa" ## [10] "4.4, 2.9, 1.4, 0.2, setosa" ## [11] "4.9, 3.1, 1.5, 0.1, setosa" ## [12] "5.4, 3.7, 1.5, 0.2, setosa" ## [13] "4.8, 3.4, 1.6, 0.2, setosa" ## [14] "4.8, 3, 1.4, 0.1, setosa" ## [15] "4.3, 3, 1.1, 0.1, setosa" ## [16] "5.8, 4, 1.2, 0.2, setosa" ## [17] "5.7, 4.4, 1.5, 0.4, setosa" ## [18] "5.4, 3.9, 1.3, 0.4, setosa" ## [19] "5.1, 3.5, 1.4, 0.3, setosa" ## [20] "5.7, 3.8, 1.7, 0.3, setosa" ## [21] "5.1, 3.8, 1.5, 0.3, setosa" ## [22] "5.4, 3.4, 1.7, 0.2, setosa" ## [23] "5.1, 3.7, 1.5, 0.4, setosa" ## [24] "4.6, 3.6, 1, 0.2, setosa" ## [25] "5.1, 3.3, 1.7, 0.5, setosa" ## [26] "4.8, 3.4, 1.9, 0.2, setosa" ## [27] "5, 3, 1.6, 0.2, setosa" ## [28] "5, 3.4, 1.6, 0.4, setosa" ## [29] "5.2, 3.5, 1.5, 0.2, setosa" ## [30] "5.2, 3.4, 1.4, 0.2, setosa" ## [31] "4.7, 3.2, 1.6, 0.2, setosa" ## [32] "4.8, 3.1, 1.6, 0.2, setosa" ## [33] "5.4, 3.4, 1.5, 0.4, setosa" ## [34] "5.2, 4.1, 1.5, 0.1, setosa" ## [35] "5.5, 4.2, 1.4, 0.2, setosa" ## [36] "4.9, 3.1, 1.5, 0.2, setosa" ## [37] "5, 3.2, 1.2, 0.2, setosa" ## [38] "5.5, 3.5, 1.3, 0.2, setosa" ## [39] "4.9, 3.6, 1.4, 0.1, setosa" ## [40] "4.4, 3, 1.3, 0.2, setosa" ## [41] "5.1, 3.4, 1.5, 0.2, setosa" ## [42] "5, 3.5, 1.3, 0.3, setosa" ## [43] "4.5, 2.3, 1.3, 0.3, setosa" ## [44] "4.4, 3.2, 1.3, 0.2, setosa" ## [45] "5, 3.5, 1.6, 0.6, setosa" ## [46] "5.1, 3.8, 1.9, 0.4, setosa" ## [47] "4.8, 3, 1.4, 0.3, setosa" ## [48] "5.1, 3.8, 1.6, 0.2, setosa" ## [49] "4.6, 3.2, 1.4, 0.2, setosa" ## [50] "5.3, 3.7, 1.5, 0.2, setosa" ## [51] "5, 3.3, 1.4, 0.2, setosa" ## [52] "7, 3.2, 4.7, 1.4, versicolor" ## [53] "6.4, 3.2, 4.5, 1.5, versicolor" ## [54] "6.9, 3.1, 4.9, 1.5, versicolor" ## [55] "5.5, 2.3, 4, 1.3, versicolor" ## [56] "6.5, 2.8, 4.6, 1.5, versicolor" ## [57] "5.7, 2.8, 4.5, 1.3, versicolor" ## [58] "6.3, 3.3, 4.7, 1.6, versicolor" ## [59] "4.9, 2.4, 3.3, 1, versicolor" ## [60] "6.6, 2.9, 4.6, 1.3, versicolor" ## [61] "5.2, 2.7, 3.9, 1.4, versicolor" ## [62] "5, 2, 3.5, 1, versicolor" ## [63] "5.9, 3, 4.2, 1.5, versicolor" ## [64] "6, 2.2, 4, 1, versicolor" ## [65] "6.1, 2.9, 4.7, 1.4, versicolor" ## [66] "5.6, 2.9, 3.6, 1.3, versicolor" ## [67] "6.7, 3.1, 4.4, 1.4, versicolor" ## [68] "5.6, 3, 4.5, 1.5, versicolor" ## [69] "5.8, 2.7, 4.1, 1, versicolor" ## [70] "6.2, 2.2, 4.5, 1.5, versicolor" ## [71] "5.6, 2.5, 3.9, 1.1, versicolor" ## [72] "5.9, 3.2, 4.8, 1.8, versicolor" ## [73] "6.1, 2.8, 4, 1.3, versicolor" ## [74] "6.3, 2.5, 4.9, 1.5, versicolor" ## [75] "6.1, 2.8, 4.7, 1.2, versicolor" ## [76] "6.4, 2.9, 4.3, 1.3, versicolor" ## [77] "6.6, 3, 4.4, 1.4, versicolor" ## [78] "6.8, 2.8, 4.8, 1.4, versicolor" ## [79] "6.7, 3, 5, 1.7, versicolor" ## [80] "6, 2.9, 4.5, 1.5, versicolor" ## [81] "5.7, 2.6, 3.5, 1, versicolor" ## [82] "5.5, 2.4, 3.8, 1.1, versicolor" ## [83] "5.5, 2.4, 3.7, 1, versicolor" ## [84] "5.8, 2.7, 3.9, 1.2, versicolor" ## [85] "6, 2.7, 5.1, 1.6, versicolor" ## [86] "5.4, 3, 4.5, 1.5, versicolor" ## [87] "6, 3.4, 4.5, 1.6, versicolor" ## [88] "6.7, 3.1, 4.7, 1.5, versicolor" ## [89] "6.3, 2.3, 4.4, 1.3, versicolor" ## [90] "5.6, 3, 4.1, 1.3, versicolor" ## [91] "5.5, 2.5, 4, 1.3, versicolor" ## [92] "5.5, 2.6, 4.4, 1.2, versicolor" ## [93] "6.1, 3, 4.6, 1.4, versicolor" ## [94] "5.8, 2.6, 4, 1.2, versicolor" ## [95] "5, 2.3, 3.3, 1, versicolor" ## [96] "5.6, 2.7, 4.2, 1.3, versicolor" ## [97] "5.7, 3, 4.2, 1.2, versicolor" ## [98] "5.7, 2.9, 4.2, 1.3, versicolor" ## [99] "6.2, 2.9, 4.3, 1.3, versicolor" ## [100] "5.1, 2.5, 3, 1.1, versicolor" ## [101] "5.7, 2.8, 4.1, 1.3, versicolor" ## [102] "6.3, 3.3, 6, 2.5, virginica" ## [103] "5.8, 2.7, 5.1, 1.9, virginica" ## [104] "7.1, 3, 5.9, 2.1, virginica" ## [105] "6.3, 2.9, 5.6, 1.8, virginica" ## [106] "6.5, 3, 5.8, 2.2, virginica" ## [107] "7.6, 3, 6.6, 2.1, virginica" ## [108] "4.9, 2.5, 4.5, 1.7, virginica" ## [109] "7.3, 2.9, 6.3, 1.8, virginica" ## [110] "6.7, 2.5, 5.8, 1.8, virginica" ## [111] "7.2, 3.6, 6.1, 2.5, virginica" ## [112] "6.5, 3.2, 5.1, 2, virginica" ## [113] "6.4, 2.7, 5.3, 1.9, virginica" ## [114] "6.8, 3, 5.5, 2.1, virginica" ## [115] "5.7, 2.5, 5, 2, virginica" ## [116] "5.8, 2.8, 5.1, 2.4, virginica" ## [117] "6.4, 3.2, 5.3, 2.3, virginica" ## [118] "6.5, 3, 5.5, 1.8, virginica" ## [119] "7.7, 3.8, 6.7, 2.2, virginica" ## [120] "7.7, 2.6, 6.9, 2.3, virginica" ## [121] "6, 2.2, 5, 1.5, virginica" ## [122] "6.9, 3.2, 5.7, 2.3, virginica" ## [123] "5.6, 2.8, 4.9, 2, virginica" ## [124] "7.7, 2.8, 6.7, 2, virginica" ## [125] "6.3, 2.7, 4.9, 1.8, virginica" ## [126] "6.7, 3.3, 5.7, 2.1, virginica" ## [127] "7.2, 3.2, 6, 1.8, virginica" ## [128] "6.2, 2.8, 4.8, 1.8, virginica" ## [129] "6.1, 3, 4.9, 1.8, virginica" ## [130] "6.4, 2.8, 5.6, 2.1, virginica" ## [131] "7.2, 3, 5.8, 1.6, virginica" ## [132] "7.4, 2.8, 6.1, 1.9, virginica" ## [133] "7.9, 3.8, 6.4, 2, virginica" ## [134] "6.4, 2.8, 5.6, 2.2, virginica" ## [135] "6.3, 2.8, 5.1, 1.5, virginica" ## [136] "6.1, 2.6, 5.6, 1.4, virginica" ## [137] "7.7, 3, 6.1, 2.3, virginica" ## [138] "6.3, 3.4, 5.6, 2.4, virginica" ## [139] "6.4, 3.1, 5.5, 1.8, virginica" ## [140] "6, 3, 4.8, 1.8, virginica" ## [141] "6.9, 3.1, 5.4, 2.1, virginica" ## [142] "6.7, 3.1, 5.6, 2.4, virginica" ## [143] "6.9, 3.1, 5.1, 2.3, virginica" ## [144] "5.8, 2.7, 5.1, 1.9, virginica" ## [145] "6.8, 3.2, 5.9, 2.3, virginica" ## [146] "6.7, 3.3, 5.7, 2.5, virginica" ## [147] "6.7, 3, 5.2, 2.3, virginica" ## [148] "6.3, 2.5, 5, 1.9, virginica" ## [149] "6.5, 3, 5.2, 2, virginica" ## [150] "6.2, 3.4, 5.4, 2.3, virginica" ## [151] "5.9, 3, 5.1, 1.8, virginica" ``` --- # Saving R objects .pull-left[ - R has a generic `.Rds` file extension for saving compressed R objects in - Good if you only need to use the data in R - Both the rio package `export` or the base `saveRDS` funtions can save any R object ```r saveRDS(object, file = "output_file.Rds") library(rio) export(object, file = "output_file.Rds") ``` ] -- .pull-right[ For tabular data, we can use: - Base R: `write.table` function - `rio` package: `export` function, which guesses the format from the output file extension - `readr` package: `write_csv`, `write_delim` ```r export(df_object, file = "my_df.csv") ``` ] --- class: inverse, center, middle # Python reading and writing --- # Accessing text files with Python - Less biology-specific packages, which simplifies the options = also less confusing. Super **easy to manipulate any kind of text**, beyond tables! - Simple `open` and `close` functions for accessing text files - `pandas` functions to access tables and many other specific file type. - Also `pickle` (binary files to be opened in Python only, can store any object in your code) and `npy`/`npz` in numpy ```python f = open("data/my_file.csv", "r"). #"r" for reading (text) first_line = f.readlines() second_line = f.readlines() rest_of_lines = f.read() f.close() f = open("data/newfile.txt", "w") # "w" for writing f.write("This will be the first line in a new file!") f.close() ``` --- # Context managers in Python - Opening and closing files as shown previously can be dangerous. - If an error occurs before the file has been `close`d, resource may never be released. - Python has context managers to deal with this problem. ```python with open("data/my_file.csv", "r") as f: #"r" for reading print(f.read()) # reads all print(f.readlines()) # contains a list where each element is a line with open("data/newfile.txt", "w") as f: #"w" for writing. Use "a" to append to an existing file f.write("This will be contained in the new file!") ``` --- # Tabular data - Can read many file types with pandas, including `.csv`, and outputs a `dataframe` ```python import pandas as pd df = pd.read_csv('data/my_file.csv') ``` - If using only numpy, can read and write `npy` files (or `npz`, the compressed version): ```python import numpy as np np.save('data/arrayfile', np.array([1,2,3])) # use 'savez' for npz np.load('data/arrayfile.npy') ``` ``` ## array([1, 2, 3]) ``` --- # Writing python objects - Use `pickle` to write and read any type of python objects, in the form of a list ```python import pickle class Person: def __init__(self, name, age): self.name, self.age = name, age p_list = [Person("John", 36), Person("Mary", 38)] with open('data/persons.pickle', 'wb') as f: # 'w' for write, 'b' for binary pickle.dump(p_list, f) with open('data/persons.pickle', 'rb') as f: persons = pickle.load(f) print(persons) ``` ``` ## [<__main__.Person object at 0x17abce210>, <__main__.Person object at 0x13880fec0>] ``` --- class: inverse, center, middle # Genomics data --- # Types of genomics data files - FASTA - Simple collections of named DNA/protein sequences - FASTQ - Extension of FASTA format, contains additional quality information. Widely used for storing unaligned sequencing reads - SAM/BAM - Alignments of sequencing reads to a reference genome - BED - Region-based genome annotation information (e.g. a list of genes and their genomic locations). Used for visualization or simple enumeration - GFF/GTF - gene-centric annotations - (big)WIG - continuous signal representation - VCF - variant call format, to store information about genomic variants --- # Reference genome as a set of coordinates .center[  ] --- # DNA Sequence data .pull-left[ ### FASTA - A sequence in FASTA format consists of: - One line starting with a ">" sign, followed by a sequence identification code. - One or more lines containing the sequence itself. ``` A --> adenosine M --> A C (amino) C --> cytidine S --> G C (strong) G --> guanine W --> A T (weak) T --> thymidine B --> G T C U --> uridine D --> G A T R --> G A (purine) H --> A C T Y --> T C (pyrimidine) V --> G C A K --> G T (keto) N --> A G C T (any) - gap of indeterminate length ``` ] .pull-right[ ### Human genome [GRCh38 Genome Reference Consortium Human Reference 38](https://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/)  ] --- # Next Generation Sequencing scheme .center[  ] .ref[https://info.abmgood.com/next-generation-sequencing-ngs-data-analysis] --- # Next Generation Sequencing scheme  .ref[https://irepertoire.com/ngs-overview-from-sample-to-sequencer-to-results/] --- # Short read data - FASTQ - An intermediate filetype that is used for downstream analysis - Usually we just pipe it to allignment software and never look at it .ref[https://knowledge.illumina.com/software/general/software-general-reference_material-list/000002211] ### General structure - `@` line with an alphanumeric ID - Base pair sequence - `+` - Quality score for each read position ``` @SIM:1:FCX:1:15:6329:1045 1:N:0:2 TCGCACTCAACGCCCTGCATATGACAAGACAGAATC + <>;##=><9=AAAAAAAAAA9#:<#<;<<<????#= ``` This is the quality score rank: ``` !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ ``` --- # Single or pair-end reads .center[  ] --- # Read depth and coverage .center[  ] .ref[https://journals.sagepub.com/doi/10.1177/1099800417750746] --- # Shotgun assembly .center[  ] .ref[https://www.differencebetween.com/difference-between-clone-by-clone-sequencing-and-shotgun-sequencing/] # Alignments - SAM and BAM --- # Some sequencing methods .center[  ] .ref[Lou, R. N., Jacobs, A., Wilder, A. P. & Therkildsen, N. O. A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. 30, 5966–5993 (2021) [10.1111/mec.16077](https://onlinelibrary-wiley-com.ezproxy.princeton.edu/share/YHRVMJNPQCIA9BPMKSJU?target=10.1111/mec.16077)] --- # Genotyping data [The Variant Call Format (VCF)](https://samtools.github.io/hts-specs/VCFv4.2.pdf)  --- # Common VCF formats ``` ##INFO=<ID=AF,Number=A,Type=Float,Description="Estimated Allele Frequencies"> ##INFO=<ID=AR2,Number=1,Type=Float,Description="Allelic R-Squared: estimated correlation between most probable ALT dose and true ALT dose"> ##INFO=<ID=DR2,Number=1,Type=Float,Description="Dosage R-Squared: estimated correlation between estimated ALT dose [P(RA) + 2*P(AA)] and true ALT dose"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=DS,Number=1,Type=Float,Description="estimated ALT dose [P(RA) + P(AA)]"> ##FORMAT=<ID=GP,Number=G,Type=Float,Description="Estimated Genotype Probability"> ``` ``` #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 127 ... 1 5385887 AX-168417668 C G . PASS AR2=1;DR2=1;AF=0.165 GT:DS:GP 0|0:0:1,0,0 ... ``` --- # Reading VCFs in R .pull-left[ ```r library(vcfR) ``` ``` ## ## ***** *** vcfR *** ***** ## This is vcfR 1.15.0 ## browseVignettes('vcfR') # Documentation ## citation('vcfR') # Citation ## ***** ***** ***** ***** ``` ```r vcf = read.vcfR("data/sample.vcf") ``` ``` ## Scanning file to determine attributes. ## File attributes: ## meta lines: 21 ## header_line: 22 ## variant count: 9 ## column count: 12 ## Meta line 21 read in. ## All meta lines processed. ## gt matrix initialized. ## Character matrix gt created. ## Character matrix gt rows: 9 ## Character matrix gt cols: 12 ## skip: 0 ## nrows: 9 ## row_num: 0 ## Processed variant: 9 ## All variants processed ``` ] --- # Reading VCFs in R ```r str(vcf) ``` ``` ## Formal class 'vcfR' [package "vcfR"] with 3 slots ## ..@ meta: chr [1:21] "##fileformat=VCFv4.0" "##fileDate=20090805" "##source=myImputationProgramV3.1" "##reference=1000GenomesPilot-NCBI36" ... ## ..@ fix : chr [1:9, 1:8] "19" "19" "20" "20" ... ## .. ..- attr(*, "dimnames")=List of 2 ## .. .. ..$ : NULL ## .. .. ..$ : chr [1:8] "CHROM" "POS" "ID" "REF" ... ## ..@ gt : chr [1:9, 1:4] "GT:HQ" "GT:HQ" "GT:GQ:DP:HQ" "GT:GQ:DP:HQ" ... ## .. ..- attr(*, "dimnames")=List of 2 ## .. .. ..$ : NULL ## .. .. ..$ : chr [1:4] "FORMAT" "NA00001" "NA00002" "NA00003" ``` --- # Using fread ```r df_vcf = fread(file='data/sample.vcf', sep='\t', header = TRUE, skip = '#CHROM') df_vcf ``` ``` ## #CHROM POS ID REF ALT QUAL FILTER ## <char> <int> <char> <char> <char> <char> <char> ## 1: 19 111 . A C 9.6 . ## 2: 19 112 . A G 10 . ## 3: 20 14370 rs6054257 G A 29 PASS ## 4: 20 17330 . T A 3 q10 ## 5: 20 1110696 rs6040355 A G,T 67 PASS ## 6: 20 1230237 . T . 47 PASS ## 7: 20 1234567 microsat1 G GA,GAC 50 PASS ## 8: 20 1235237 . T . . . ## 9: X 10 rsTest AC A,ATG 10 PASS ## INFO FORMAT NA00001 NA00002 ## <char> <char> <char> <char> ## 1: . GT:HQ 0|0:10,10 0|0:10,10 ## 2: . GT:HQ 0|0:10,10 0|0:10,10 ## 3: NS=3;DP=14;AF=0.5;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51 ## 4: NS=3;DP=11;AF=0.017 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3 ## 5: NS=2;DP=10;AF=0.333,0.667;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2 ## 6: NS=3;DP=13;AA=T GT:GQ:DP:HQ 0|0:54:.:56,60 0|0:48:4:51,51 ## 7: NS=3;DP=9;AA=G;AN=6;AC=3,1 GT:GQ:DP 0/1:.:4 0/2:17:2 ## 8: . GT 0/0 0|0 ## 9: . GT 0 0/1 ## NA00003 ## <char> ## 1: 0/1:3,3 ## 2: 0/1:3,3 ## 3: 1/1:43:5:.,. ## 4: 0/0:41:3:.,. ## 5: 2/2:35:4:.,. ## 6: 0/0:61:2:.,. ## 7: 1/1:40:3 ## 8: ./. ## 9: 0|2 ``` --- # Gene expression .center[  ] --- # Gene expression .center[ 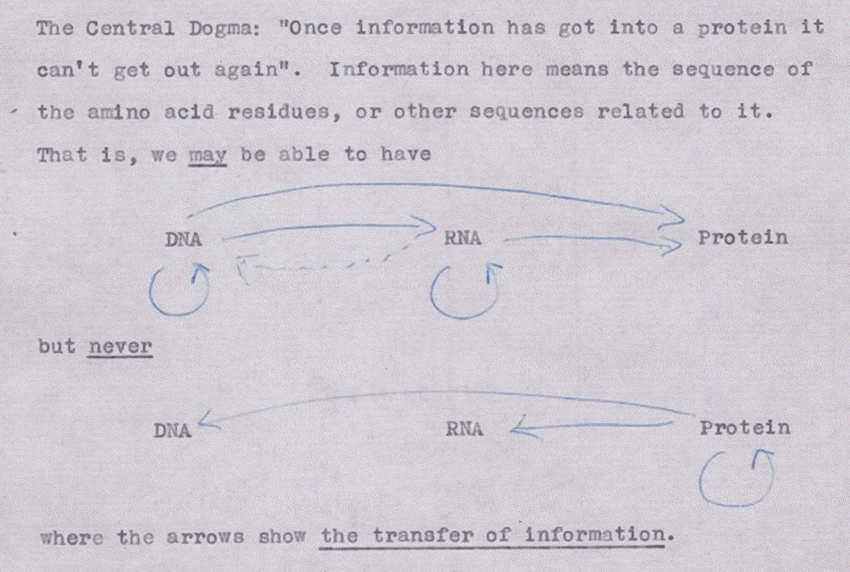 ] --- # Gene expression 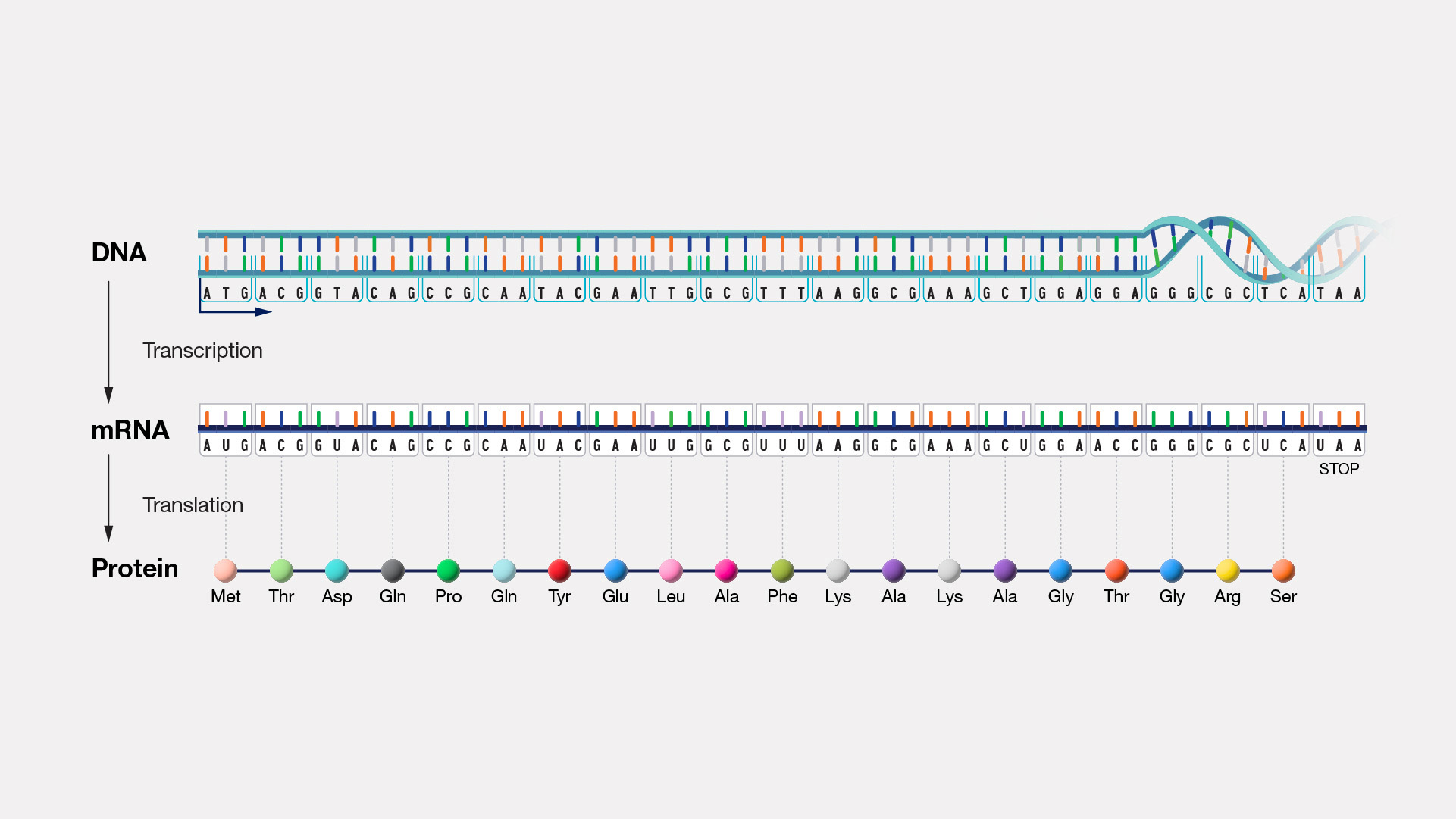 --- # Gene expression data (RNAseq) .pull-left[  .ref[https://www.nature.com/articles/nrg2484] ] .pull-right[ - Aligned RNA reads that map to genes are contributed - The counts are proportional to RNA expression ```r counts <- read.delim("https://raw.githubusercontent.com/ucdavis-bioinformatics-training/2018-June-RNA-Seq-Workshop/master/thursday/all_counts.txt") counts[1:10, 1:7] ``` ``` ## C61 C62 C63 C64 C91 C92 C93 ## AT1G01010 289 317 225 343 325 449 310 ## AT1G01020 127 78 142 130 156 146 144 ## AT1G03987 0 0 0 0 0 0 0 ## AT1G01030 17 25 32 24 22 25 21 ## AT1G01040 605 415 506 565 762 854 658 ## AT1G03993 1 1 0 0 0 0 1 ## AT1G01046 0 0 0 0 0 0 0 ## ENSRNA049757489 0 0 0 0 0 0 0 ## AT1G01050 1164 876 935 979 1146 1254 948 ## AT1G03997 0 0 0 0 0 0 0 ``` ]